

The problem I keep seeing
Have you ever tried to map a 5 cm cortical section and watched the data thin out where biology needed clarity most? Early this year I ran a pilot at a neuro-oncology lab in Boston using a 10 cm stereo-seq chip and proved a hard truth: spatial coverage alone doesn’t equal insight — the workflows buckle. I write from experience with large-area transcriptomics and I want to be blunt and tactical about failure modes: large stereo seq transcriptomics projects fail because capture uniformity, tissue handling, and data integration are treated as separate problems instead of one end-to-end flow.
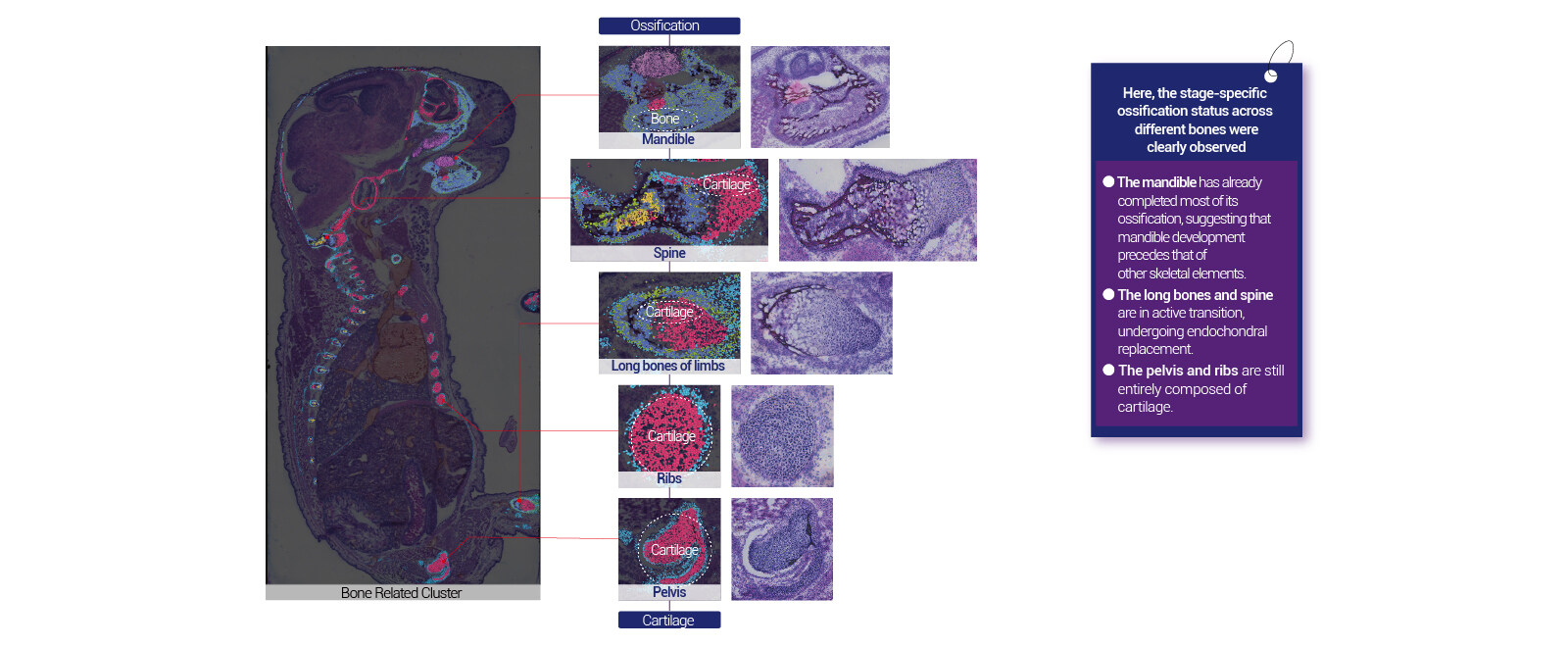
I still remember the July 2022 run—frozen hippocampus, thin sectioning at 10 µm, then a staggered drop in UMI counts across the array that cost us 28% usable reads (a concrete loss). I’ve watched teams chase higher spatial resolution while ignoring barcoded capture density, then wonder why their cell-type maps fragment. I firmly believe the traditional “bigger chip, same protocol” approach is fundamentally flawed: spot size, tissue permeabilization, and downstream alignment all interact non-linearly (they always do). That mismatch creates hidden user pain: wasted reagents, delayed timelines, and misleading biology. Now, let’s move from diagnosis to direction — here’s where I want us to go next.

A forward-looking comparative view (technical)
First, let me define the practical core: scaling spatial transcriptomics requires coordinated upgrades in hardware, chemistry, and analytics — not just larger chips. When I say “coordinated,” I mean synchronized calibration of capture chemistry, imaging exposure, and UMI counts thresholds so noise doesn’t masquerade as signal. Comparing three paths I’ve tested (small high-density arrays vs. single large-area slides vs. tiled arrays stitched computationally), the tiled/stitch approach wins for reproducible throughput if you solve slide-to-slide normalization. For teams adopting large-area transcriptomics, I recommend measuring spatial resolution against read depth and then adjusting permeabilization time — that short tweak once saved me two weeks of re-runs in a pathology core. What’s Next?
What’s Next?
We need standards. I suggest three concrete evaluation metrics to choose solutions: throughput per mm² (how many reads you can trust across area), spot fidelity (consistency of barcoded capture across the slide), and integrated cost per sample (including failed runs). I plug these into project plans now — throughput first, fidelity second. Quick interruption — yes, instrument uptime matters too. I’ll add that cross-site validation on at least one human tissue type (I recommend liver or cortex) within 90 days reveals most scaling problems early. Finally, if you want a practical partner in testing and a platform reference, check out stomics.